How to qualify, classify and CE mark software
Medical devices shall have CE mark before being sold in the EU. The process to have CE mark can be summarized this way:
- Determining the class of the device,
- Choosing the CE procedure to apply,
- Declaring CE conformity of the device.
Software follows exactly the same process as other devices. Here are the steps to follow to CE mark software.
This page is the Medical Device Regulation (MDR) version.
Go to the Medical Device Directive (MDD) version
MDR CE Marking
I used the references below to create this article:
- Consolidated version of MDR
- Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR: MDCG 2019-11
- The lastest version of the Manual on borderline and classification of medical devices on this page, section "borderline devices"
- Summary list of titles and references harmonised standards under Directive 93/42/EEC for Medical devices and the Draft list of harmonized standards (as long as there is no list for MDR)
Qualifying software as medical device
The first question to ask is whether software is a medical device or not.
The answer to this question is all based on its intended use. It shall be for diagnostic, treatment, prognosis, prevention purposes. It is stated in the definition of medical device in article 2 of the MDR.
The MDCG 2019-11 ref.2 has two decision trees that are an help to qualify software as medical device or in-vitro diagnostic device. The guidance contains also a comprehensive list of examples in annex 1 and 2, with types of software which qualify or don't qualify as medical devices.
Not medical devices
To give the most recurring examples of software, which are not medical device, we have:
- Systems managing administrative patient data, like appointment scheduling, billing,
- Prescription Management Systems (but drug dose calculators using patient data like weight or body mass index are medical devices),
- Systems storing patient data, without data processing,
- Radiological Information System (RIS).
These software are not medical devices, thanks to the decision tree found in MDCG 2019-11. The most common situation is to answer "Yes" to the Decision step 3 of that guide.
Warning: these types of software are not qualified as medical devices in EU. But they may qualify as medical devices in other regions of the world, especially in the US.
Borderline cases
However, even with the help of the guidances, it may be difficult to qualify software as medical device. Borderline cases exist, usually with standalone software, like software that could be used at the same time for general wellness purposes or for treatment purposes. In this case, the qualification of medical device stems from the claims in the intended use.
For example:
- The intended use of a mobile app claims that it records and computes statistics about what the user eats and the quantities of carbs, fat, etc. It is for general wellness purposes, it is not a medical device.
- The intended use of the same mobile app (technically) now claims that it records and computes statistics about what the user eats and the quantities of carbs, fat, for diabetes management. It is for management of diabetes - an aid to the treatment of a disease - it is a medical device.
The Borderline Manual ref.3 is also a good source of information to determine whether software is a medical device or not.
Medical devices and intended use
The intended use is the responsibility of the manufacturer. Having an intended use in the scope of medical devices or not can also be seen as a management decision. Thus, same software could be qualified as medical device or not, depending on management decision on what will be claimed with that software!
However, there are cases when software is medical device given its use or its context of use:
- Software, which is an accessory of a medical device, is a medical device,
- Software, which drives of influence the use of a medical device is a medical device.
And there are cases where the manufacturer can't hide behind the intended use they wrote, to escape from the scope of the medical device regulation.
Imagine that a manufacturer sells software with an intended use for general wellness. But then promotes its software for diagnostic or treatment purposes or for use combined with a medical device in such a way that it can be qualified as an accessory of the device. Then it is by real use a medical device, even if it's not claimed in the intended use.
Qualifying software as a medical device can be a difficult task hence the result has heavy consequences in terms of time and money, and in responsibility.
The guidances quoted above may not have enough information for peculiar cases. Thus it's worth contacting a notified body or a competent authority to assess the status of your software.
Modular Approach
If you have a software platform delivering various services, qualifying one subset of these services as medical device doesn't qualify the whole platform as a software medical device. Fortunately, section 7 of the MDCG 2019-11 allows software editors to apply a modular approach.
Only the modules containing the functions with a medical purpose will be qualified as medical devices. The rest of the software platform will remain health software outside the scope of the MDR. The condition to have such status is to ensure a architectural segregation between medical devices and non-medical device components.
Make use of the modular approach, as soon as you have a platform with several modules. It will save time and money to exclude some parts of that platform form the MDR scope.
Classifying software
Each medical device shall be classified in one class: class I, class IIa, class IIb or class III. It depends on the level of risk generated by the use of the device. This is done by reviewing the rules in the annex VIII of the MDR ref.1: Annex VIII, section II.3.3 states that
- If the software is independent of any other device, it shall be classified in its own right., this is the rule for standalone software,
- Software, which drives a device or influences the use of a device, shall fall within the same class as the device., this is the rule for software connected to another device, hardware or software.
The terms “drives or influence” may be interpreted. Here are some examples:
- Software with functions modifying the state of the device actually drives it,
- Software, which only displays data collected from the device, may not be at first sight seen as “driving” the device. But if the user uses data provided by the software to modify the state of the device, then the software “influences” the device.
- Software, which only collects data for archiving purpose doesn’t influence the medical device and may be classified alone.
Rule 1
Just for information, the rule 1, applicable to devices not in contact with a patient, is always applicable to standalone software. That's not a very useful rule, as is states that such device is in class I. This rule is always overtaken by other rules discussed below.
Rules 9 to 13 of the MDR
Standalone software is considered as an active medical device (powered by electricity. No current, no software). Active medical devices are governed by rules 9 to 13. The purpose of the MDCG 2019-11 guidance ref. 2 is to give some explanations on how to interpret these rules.
The table below gives the interpretations of each rule for software, based on section 4.2 of MDCG 2010-11 ref. 2 and section 9 of the Borderline Manual ref. 3.
| Rule # | Text | Software |
|---|---|---|
| 9 | All active therapeutic devices intended to administer or exchange energy | Devices classified by this rule are mostly electrical equipment used in surgery such as lasers and surgical generators. In addition there are devices for specialized treatment such as radiation treatment. Another category consists of stimulation devices, although not all of them can be considered as delivering dangerous levels of energy considering the tissue involved. Standalone software intended to control or monitor such devices, or directly influence their performance, are also classified by this rule. |
| 10 | Active device for diagnosis | This primarily covers a whole range of widely used equipment in various fields, e.g. ultrasound diagnosis, capture of physiological signals and therapeutic and diagnostic radiology. Vital physiological processes and parameters include, for example respiration, heart rate, cerebral functions, blood gases, blood pressure and body temperature. Standalone software intended to control or monitor such devices, or directly influence their performance, are also classified by this rule. See also paragraph about PACS below. |
| 11 | This is The Rule for standalone software | This rule is intended to cover standalone software. It usually overpasses all other classification rules. See below the discussion on this rule. |
| 12 | Active devices to administer or remove medicine | This rule is intended to primarily cover drug delivery systems and anesthesia equipment. Standalone software intended to control or monitor such devices, or directly influence their performance, are also classified by this rule. |
| 13 | All other active devices | This is a fallback rule to cover all active devices not covered by the previous rules. Class I stand alone software can also come with a measuring function. |
Some examples:
Rule 9:
- Software embedded in a muscle stimulator, an incubator, a laser …
- Software connected through network (wired or wireless) to a therapeutic device. Eg: remotely monitored physiotherapy equipments,
- Radiotherapy planning system used to calculate the dose of ionizing radiation to be administered to the patient, insulin dosage planning stand alone software.
Rule 10:
- Software embedded in any scanner (ultrasound, X-Ray, MRI), an electronic thermometer …
- Software connected through network (wired or wireless) to a diagnostic device. Eg: Picture Archiving and Communication System (PACS),
Rule 11: Almost all kind of standalone software with diagnostic, monitoring, therapeutic purpose,
- Standalone software processing patient data to allow direct diagnostic. Eg: a web app used to compute and display diagnostic data.
- Standalone software processing patient data as an aid to diagnostic. Eg: a mobile app used to compute a score.
- Standalone software connected to a multifunction monitor, used to remotely monitor the parameters.
Rule 12:
- Software embedded in infusion pumps, ventilators, nebulizers, suction pumps, dialysis equipment,
- Software connected through network (wired or wireless) to a device delivering or administering medicine,
Rule 13: Anything else
- Software embedded in an hospital bed
- Software used to monitor or control a class I device
- All other standalone software. Use this rule with economy, when every other rule has been rejected after extensive interpretation. It may be attractive to have software on smartphones or tablets fall in rule 13. This is often not the case. Rule 11 is the name of the game.
Special considerations for Picture Archiving and Communication Systems:
Classification rules cannot genuinely cover every case. The working group on “borderline and classification” was created to give recommendations on such cases. The manual on borderline classification of medical devices V1.22, was published in the good old times of the EC directive. A chapter deals with Picture Archiving and Communication System (PACS). PACS shall be class IIb, IIa or class I, depending on their purpose. Here are some examples. If the PACS:
- Stores images and doesn’t modifies them, then it is class I,
- Is used to monitor and control the scanner, then it has the class of the device,
- Is a post-processing tool used to interpret raw images, then it is class IIa.
This guide was edited for the directive. Its content looks still applicable with the MDR. Namely, the discussion on PACS outcomes is consistent with the classification of MDR rule 11.
Rule 11
The verbatim of the rule is given below:
- Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
- death or an irreversible deterioration of a person's state of health, in which case it is in class III; or
- a serious deterioration of a person's state of health or a surgical intervention, in which case it is classified as class IIb.
- Software intended to monitor physiological processes is classified as class IIa,
- except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
- All other software is classified as class I.
The MDR definition of medical device contains the words diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease. The rule 11 places software in class IIa when it is intended to provide information which is used to take decisions with diagnosis or therapeutic purposes
Diagnosis, monitoring, prediction, prognosis, treatment, alleviation are diagnosis or therapeutic purposes. There remains only prevention outside the scope of rule 11. Thus, there is 99% chance that your software is in class IIa. The MDCG 2019-11 doesn't say the contrary.
The annex II of MDCG 2019-11 provides an interpretation of rule 11 with the following table (ugly explicit colors added by me):
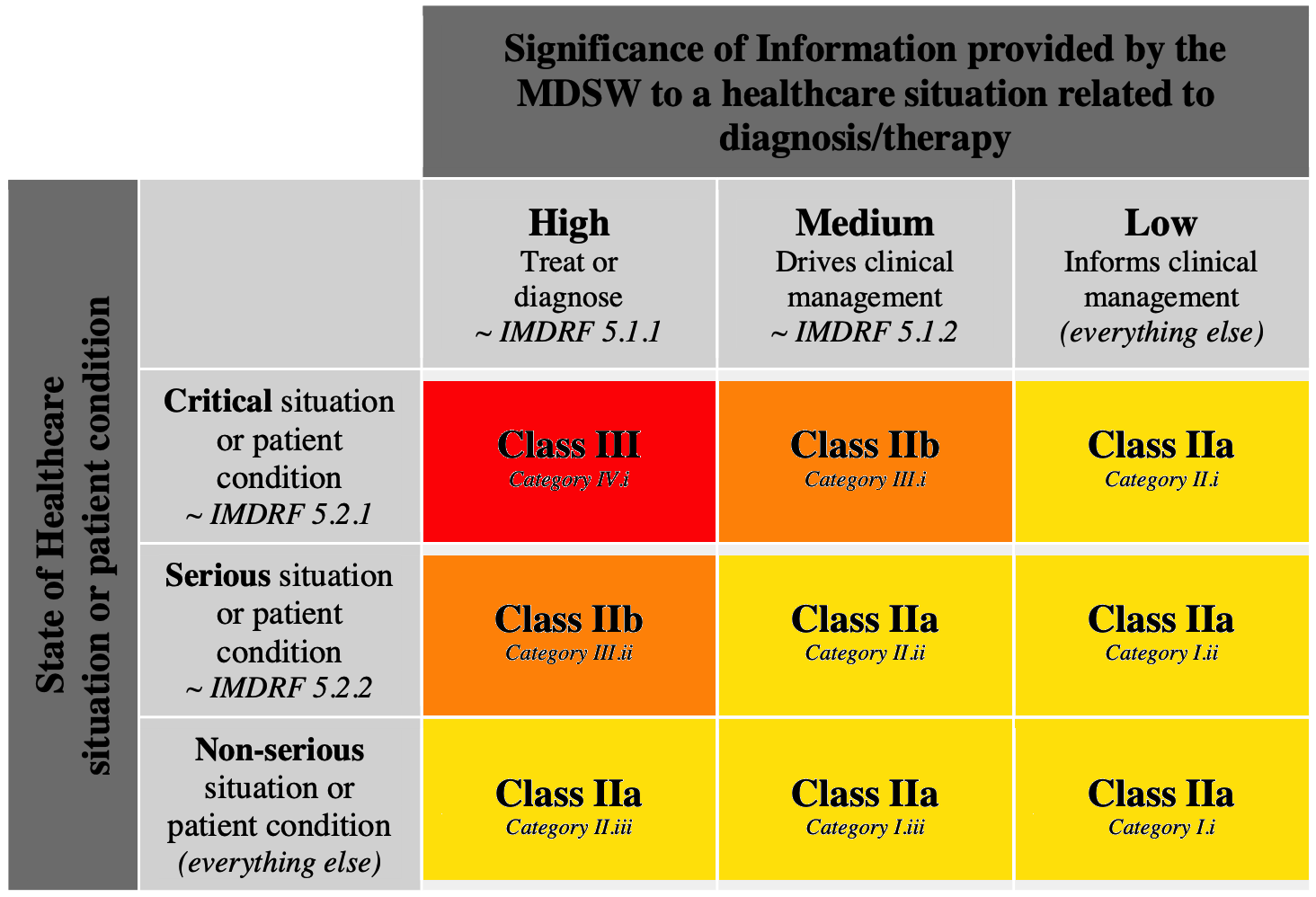
It reduces the probability to place your software in a class higher that class IIa, by combining the impact of the information provided by software with the patient state.
The definitions of significance of information and state of health come from another guidance published outside EU: IMDRF/SaMD WG/N12 FINAL:2014 Software as a Medical Device: Possible Framework for Risk Categorization and Corresponding Considerations.
Here are these definitions, copied from the IMDRF document:
Significance of information:
To treat or to diagnose
Treating and diagnosing infers that the information provided by the SaMD will be used to take an immediate or near term action:
- To treat/prevent or mitigate by connecting to other medical devices, medicinal products, general purpose actuators or other means of providing therapy to a human body
- To diagnose/screen/detect a disease or condition (i.e., using sensors, data, or other information from other hardware or software devices, pertaining to a disease or condition).
To drive clinical management
Driving clinical management infers that the information provided by the SaMD will be used to aid in treatment, aid in diagnoses, to triage or identify early signs of a disease or condition will be used to guide next diagnostics or next treatment interventions:
- To aid in treatment by providing enhanced support to safe and effective use of medicinal products or a medical device.
- To aid in diagnosis by analyzing relevant information to help predict risk of a disease or condition or as an aid to making a definitive diagnosis.
- To triage or identify early signs of a disease or conditions.
To Inform clinical management
Informing clinical management infers that the information provided by the SaMD will not trigger an immediate or near term action:
- To inform of options for treating, diagnosing, preventing, or mitigating a disease or condition.
- To provide clinical information by aggregating relevant information (e.g., disease, condition, drugs, medical devices, population, etc.)
Healthcare Situation:
Critical situation or condition
Situations or conditions where accurate and/or timely diagnosis or treatment action is vital to avoid death, long-term disability or other serious deterioration of health of an individual patient or to mitigating impact to public health. SaMD is considered to be used in a critical situation or condition where:
- The type of disease or condition is:
- Life-threatening state of health, including incurable states,
- Requires major therapeutic interventions,
- Sometimes time critical, depending on the progression of the disease or condition that could affect the user’s ability to reflect on the output information.
- Intended target population is fragile with respect to the disease or condition (e.g., pediatrics, high risk population, etc.)
- Intended for specialized trained users.
Serious situation or condition
Situations or conditions where accurate diagnosis or treatment is of vital importance to avoid unnecessary interventions (e.g., biopsy) or timely interventions are important to mitigate long term irreversible consequences on an individual patient’s health condition or public health. SaMD is considered to be used in a serious situation or condition when:
- The type of disease or condition is:
- Moderate in progression, often curable,
- Does not require major therapeutic interventions,
- Intervention is normally not expected to be time critical in order to avoid death, long- term disability or other serious deterioration of health, whereby providing the user an ability to detect erroneous recommendations.
- Intended target population is NOT fragile with respect to the disease or condition.
- Intended for either specialized trained users or lay users.
Note: SaMD intended to be used by lay users in a "serious situation or condition" as described here, without the support from specialized professionals, should be considered as SaMD used in a "critical situation or condition".
Non-Serious situation or condition
Situations or conditions where an accurate diagnosis and treatment is important but not critical for interventions to mitigate long term irreversible consequences on an individual patient's health condition or public health. SaMD is considered to be used in a non-serious situation or condition when:
- The type of disease or condition is:
- Slow with predictable progression of disease state (may include minor chronic illnesses or states),
- May not be curable; can be managed effectively,
- Requires only minor therapeutic interventions, and
- Interventions are normally noninvasive in nature, providing the user the ability to detect erroneous recommendations.
- Intended target population is individuals who may not always be patients.
- Intended for use by either specialized trained users or lay users.
Alas, no matter what you do with these rules, your software will be in class IIa or higher.
Measuring Function
On top of the class, you shall also determine if your software has a measuring function. This is explained in the MDCG 2021-24, section 3.1.6. If your software matches the all three criteria in the guide, then your software has a measuring function. Thus, you have to validate it in your design file.
Choosing the conformity assessment procedure to apply
The MDR defines several procedures to CE mark a device. They are described in article 51 of the MDR, combined with article 61 for clinical evaluation, and articles 83 to 87 for post-market surveillance.
The principle of these procedures is to build a quality management system and build technical file containing data about the medical devices. The conformity assessment procedure to apply differs with the class of the medical device. Here are the possible combinations for standalone software:
| Class# | QMS Assessment | Technical File Assessment | Clinical Investigation | Post-Market Surveillance |
|---|---|---|---|---|
| III | Annex IX QMS Chapters I, III | Annex IX Chapter II Technical Documentation for every device | Mandatory | Updated every year and sent to Notified Body |
| Annex XI – Part A Production Quality Assurance | Annex X Type Examination | |||
| IIb | Annex IX QMS Chapters I, III | Annex IX Chapter II Technical Documentation per generic device group | Not mandatory, but difficult to avoid with innovative functions | Updated every year |
| Annex XI – Part A Production Quality Assurance | Annex X Type Examination | |||
| IIa | Annex IX QMS Chapters I, III | Annex IX Chapter II Technical Documentation per device category | Not mandatory, but difficult to avoid with innovative functions | Updated every two years |
| Annex XI – Part A Production Quality Assurance | Annex II and Annex III Technical Documentation Assessed per device category | |||
| Im (with measuring function | Annex IX QMS Chapters I, III | Annex IX Chapter II Technical Documentation per device category | Not mandatory | Updated as appropriate |
| I (just in case...) | None, apply Article 10 | Self declaration of conformity of Annex II and Annex III Technical Documentation |
The Annex XI – Part B Product Verification is not suitable for software, as the product verification requires to examine every device individually. Annex XI – Part B targets hardware devices, not standalone software.
Characteristics of standalone software
- Standalone software by essence doesn't enter into contact with a patient, isn't implantable, doesn't contain substances, and isn't combined with drugs. Thus, conformity assessment procedures don't require a second review of technical documentation by subject matter experts after the review by the notified body.
- Software is also characterized by the absence of categories or generic groups (e.g. implants with several sizes). When a software editor has several software, they are marketed individually or as a software suite, each software in the suite being a single product (think about MS Office).
- The Harmonized Standards (see ref. 4) for software are IEC 62304, IEC 62366-1 and section 14 of IEC 60601-1. We can also add IEC 82304-1 and IEC 81001-5-1, not harmonized yet but recognized by the FDA and under the radar of Notified Bodies. All five standards require manufacturers to have a design control procedure for software.
- Software is very prone to be updated every year or even every quarter, with major changes to existing functions or new functions. Every year is quite a good frequency. Trying to update software at a higher pace is a difficult task, not controlled by the editor. The Notified Bodies impose their rhythm.
The consequence of these specific traits is to have very few differences between class IIa and class IIb software:
- If software contains innovative functions, it will require clinical data, usually coming from a clinical investigation on that software (and not a competitor or some research version), no matter the class,
- Every software is different, generic groups and categories aren't meaningful. Thus, every technical file will be reviewed by the Notified Body, no matter the class. Unless you have a very long list of software, something quite uncommon,
- If software is updated on a yearly basis with major changes (something common for standalone software), it will require an annual post-market surveillance report (PSUR), no matter the class,
- There are differences of sampling plans for class IIa and class IIb, after the initial conformity assessment. If you update your software every year, this sampling plan isn't relevant, since the Notified Body will have to review the changes, no matter the class.
All of this is also true for class III software, with some more overhead on the review of the clinical investigation and clinical evaluation by the Notified Body and group of clinical experts.
Annex IX or Annex XI?
The main difference between Annex IX and annex XI is linked to the way manufacturer management organizes the company. Usually:
- Annex IX is applied to the whole company, for every product in the scope of the Quality Management System. The company is ISO 13485 certified and most of them have an activity in medical devices only,
- Annex X, and XI can be applied to a subset of products delivered by the company. It may be ISO 9001 certified for all its products. A subset of products is composed of medical devices, for which the company chose to apply the relevant annexes of the MDR,
- Annex X and XI can also be chosen by a company with a single product or a single family of products.
Of course, every case may be found in the nature, like a company manufacturing only medical devices and applying other annexes than annex IX. But this is more and more frequent with the MDR to have software editors with a single product, which are ISO 13485 certified, and apply annex IX.
A word on procedures after design
Design control procedure is of high importance. But other tasks shall not be neglected.
Annex XI is all about production. For software, production is made of installation, configuration, testing:
- Installation: flashing the software on a micro controller, installation on a server, on clients, etc…
- Configuration: defining the right configuration files for the customer, using administrative tools,
- Testing: test of the final configuration defined for the customer; test of software integrated in a product item; routine security tests in production …
These steps shall be monitored very carefully. Some may be done by the customer and shall be double-checked. A divergence from expected behavior is very common with software. It’s mandatory to have procedures to handle these tasks to have software work properly.
Bug fixes and enhancements: A full quality system according to annex IX shall have procedures to manage bugs and enhancements. So do procedures according to annex XI. Collecting bug and enhancements is a mandatory task, based on the requirements of MDR article 10.
The IEC 62304 standard gives a comprehensive list of processes to implement in order to have software developed and maintained according the state of the art. Applying this standard is the best thing to do prior to any software medical device CE conformity project.
Declaring CE conformity of the device
Depending on the class of the device, a notified body issues a certificate of conformity. then the manufacturer declares the conformity:
- Step 1 for Class I (sterile or with a measuring function), Class IIa, Class IIb and Class III devices: the notify body delivers a certificate of conformity,
- Step 2: the manufacturer declares the conformity.
Of course, the notified body doesn’t establish the certificate of CE conformity in a snap of a finger. It reviews the whole documentation produced about the device. The process can take up to a year (yes, A YEAR). But if the documentation follows clearly a software design process compliant with the standards quoted above, then the job of the notified body is made easier. It should prevent them from asking troubleshooting questions about software and/or issuing non-conformities.
Conclusion
Congratulations if you read this till the end!
Placing software medical device on the European market is a long journey with lots of obstacles and long detours.
I hope you know a bit more about it, at the end of this page.
First version: August 2020
Updated on March 2024
Reason: periodic review of articles
Added measuring function
Fixed dead links and updated guidance info.
Published on Wednesday, 5 August 2020 by Mitch